M. Rath, L. Pauling
Journal of Orthomolecular Medicine 1991, 6:125-134
Summary
Human cardiovascular disease (CVD) is the result of the accumulation of lipoprotein(a), Lp(a), rather than of low density lipoprotein (LDL), in the vascular wall. It is generally not the consequence of plasma LDL levels, but rather of the level of Lp(a), which is formed in the liver in amounts largely determined by the rate of synthesis of apo(a). This rate is increased by low ascorbate concentrations. Human CVD is primarily a degenerative disease caused by ascorbate deficiency. This deficiency is the result of the inability of humans to synthesize endogenous ascorbate combined with an insufficient dietary ascorbate intake. The deficiency is aggravated by genetic defects such as the LDL receptor defect and by exogenous risk factors for CVD leading to additional ascorbate depletion. Ascorbate deficiency results in morphologic changes of the vascular wall.
In order to avoid the fatal consequences of extreme ascorbate depletion, such as hemorrhagic bleeding in scurvy, ascorbate deficiency simultaneously increases the plasma concentration of vasoconstrictive and hemostatic risk factors, including Lp(a) and fibrinogen. Chronic ascorbate deficiency leads to the extracellular accumulation of Lp(a) and fibrinogen/fibrin, the hallmarks of the atherosclerotic lesion. The underlying impairment of the vessel wall is unmasked mainly at sites of altered hemodynamic conditions, leading to myocardial infarction and stroke as the predominant manifestations of human CVD. Thus for patients with coronary or cerebrovascular disease the instability of the vessel wall due to ascorbate deficiency is the leading risk factor, rather than plasma constituents. In contrast, risk factors in plasma trigger the manifestation of peripheral vascular disease (PVD). In this condition plasma constituent such as oxygen free radicals from cigarette smoke or oxidatively modified triglyceride-rich lipoproteins exert a noxious effect on the vascular wall in the periphery and PVD develops. Ascorbate depletion of the vascular tissue is also a precondition for the manifestation of PVD. Human CVD is multifactorial. Ascorbate deficiency, however, is the common denominator of this disease. The comprehensive pathogenetic and therapeutic concept presented in this paper represents the solution to the puzzle of human cardiovascular disease and should lead to the improvement of human health.
Full Study
"My dear Kepler, what do you say of the leading philosophers here to whom I have offered a thousand times of my own accord to show my studies, but who, with the lazy obstinacy of a serpent who has eaten his fill, have never consented to look at the planets, or moon, or telescope? Verily, just as serpents close their eyes, so do men close their eyes to the light of truth."
Galileo Galilei in a letter
to Johannes Kepler ca. 1630
Introduction
We recently formulated the concept that lipoprotein(a), Lp(a), is a surrogate for ascorbate, vitamin C. (1) This concept revealed the physiological role of Lp(a) as well as new therapeutic approaches. On the basis of earlier work and additional experimental and clinical evidence we now present a detailed theory of human CVD. The primary cause of human CVD is a deficiency in ascorbate leading to the deposition of Lp(a) and fibrinogen/fibrin in the vascular wall. We elucidate the interaction of ascorbate and Lp(a) and present a pathomechanism that differs from existing concepts (2,3,4) in that it is able to explain the unique features of human atherosclerosis. We also present prophylactic and therapeutic considerations that open new pathways to prevention and treatment of CVD.
The Pivotal Role of Lp(a) in Human Cardiovascular Disease
Lp(a) was discovered by Kare Berg in 1963. (5) It is closely similar to LDL, the main difference being that a glycoprotein, apo(a), is attached by a disulfide bond to the apoprotein of LDL, apo B, giving a larger surface area to the lipoprotein sphere. The c-DNA sequence of apo(a) shows a striking homology to that of plasminogen (6), with multiple repeats of kringle 4, one kringle 5 and a protesase domain. Because of the homology of apo(a) with plasminogen Lp(a) has been called the missing link between atherogenesis and thrombogenesis (7).
Evidence that Lp(a), not LDL, is the primary lipoprotein responsible for initiating the development of atherosclerosis was reported by one of us and his colleagues at Hamburg University (8,9,10). In the most comprehensive studies assessing the role of Lp(a) in human vascular wall yet reported it was found that Lp(a), not LDL accumulates selectively in the vascular wall of CVD patients. Moreover the extracellular accumulation of Lp(a) was closely correlated to the development of atherosclerotic plaques.
Most importantly, in several hundreds of histological cross sections from the human coronary arteries and the aorta immunostaining for apoB, without congruent staining for apo(a) was a rare event, indicating that the vascular wall deposition of LDL alone occurs rarely (9). The deposition of Lp(a) in the vascular wall as determined by immuno-morphometric analysis because extraction methods overestimate the role of LDL: a major fraction of Lp(a) is found dissociated in the vascular wall into apo(a) and the LSDL-like particle particularly under post-mortem conditions. (8) Earlier investigators have evidently failed to differentiate between LDL and Lp(a) so that the initiation of atherosclerotic lesions was incorrectly attributed to LDL.
This conclusion was recently confirmed by a study determining plasma risk factors in patients with inherited LDL-receptor defects. In these familial hypercholesterolemic patients the incidence of CVD was significantly determined by the Lp(a) plasma concentration, with total cholesterol and LDL cholesterol in plasma not related to the clinical manifestation of CVD.
There is now strong clinical and experimental evidence that Lp(a) is a more important risk factor than total cholesterol or LDL-cholesterol for coronary heart disease (12), stroke (13), as well as restenosis of vein grafts after coronary bypass surgery (14). We therefore conclude that Lp(a) is the lipoprotein primarily responsible for the initiation of human CVD. The role of LDL is best characterized as an aggravating risk factor for CVD in patients with simultaneously elevated Lp(a) plasma levels.
The Ascorbate-Lp(a) Connection
We observed that Lp(a) has mainly been detected in the plasma of man, other primates and a few other species that have lost the ability to synthesize ascorbate and consequently have low ascorbate levels compared to animals with endogenous ascorbate production. We do not exclude, however, that small amounts of Lp(a) will also be found in other species. The loss of ascorbate synthesis is the result of a genetic mutation in the gene for L-gulono-c -lactone oxidase; this mutation occurred 40 million years ago in an ancestor of the primates. Subsequently, Lp(a) became a major plasma constituent in primates and man. We therefore proposed that Lp(a) is a surrogate for ascorbate. Vice versa, ascorbate is a surrogate for Lp(a), since in most species Lp(a) is replaced by ascorbate without any disadvantage.
Previously, it has been assumed that Lp(a) is primarily a pathogenic particle and that Lp(a) plasma concentrations are primarily determined by genetic factors. Our publication of the Lp(a)-ascorbate connection marked a turning point in research directions and suggested numerous investigations. Subsequently, it was shown that ascorbate, the strongest reducing agent normally present in the body, and also synthetic reducing agents such as N-acetylcysteine (15), decrease Lp(a) plasma levels. In a clinical trial in CVD patients an increased intake of ascorbate lowered the plasma Lp(a) level (unpublished observations).
Moreover, we proposed that Lp(a) strengthens the vascular wall, particularly in ascorbate deficiency. At low ascorbate concentrations the synthesis of collagen and elastin is impaired and the deposition of Lp(a) helps to control the resulting instability of the vessel wall and to contain disease progression. Apo(a), a macromolecule, would compensate for this impairment and its demonstrated binding to glycosaminoglycans and other compounds of the extracellular matrix would be beneficial. Moreover, apo(a) has been shown top bind with high affinity to proline and hydroxyproline and is likely to bind to collagen and elastin, macromolecules that are enriched in these amino acid residues. Increased intake of ascorbate eliminates the need for Lp(a) to strengthen the blood vessels and thus ascorbate can replace Lp(a).
We have recently been able to confirm that ascorbate can replace Lp(a) at the site of the disease process. In this pilot study we used the hypoascorbemic guinea pig , an animal like man, unable to synthesize ascorbate but able to synthesize apo(a). When fed dietary ascorbate in small amounts, corresponding approximately to the usual human intake, these animals rapidly develop atherosclerotic plaques and deposit Lp(a) in the vascular wall. Larger intakes of ascorbate inhibited the deposition of Lp(a) in the arterial wall and prevented the development of atherosclerosis. (16)
Ascorbate and the Regulation of Plasma Lp(a)
Lp(a) plasma levels among individuals vary by as much as 1000 fold. This considerable variation is to a large extent the result of genetic factors determining the synthesis of apo(a), but also those of apoB and lipids. It may be that the modifying genes controlling apo(a) synthesis at the optimum level have not yet become fully effective, so that in some individuals this synthesis has overshot the mark, predisposing them to CVD.
Beside genetic factors, Lp(a) plasma concentrations are also regulated by dietary factors, one of them being niacin, which has been shown to lower plasma Lp(a) levels (17). Another dietary factor is ascorbate. We have obtained preliminary results that ascorbate decreases apo(a) synthesis in human hepatoma cells in vitro. Ascorbate may also decrease the assembly of the Lp(a) particle by reducing the disulfide formation between apo(a) and apo B in the liver.
Ascorbate Defiency, the Risk Profile for CVD and Lp(a)
Ascorbate depletion is the common metabolic denominator of endogenous and exogenous risk factors for CVD. Many genetic defects are associated with ascorbate deficiency. As a result of a genetic defect the rate-constants of certain enzyme-controlled metabolic reactions are decreased. These rate constants can be increased towards normal values by increasing the concentrations of certain cofactors (18). In the attempt to normalize these decreased rate constants, ascorbate and other essential cofactors for metabolic reactions are depleted. Ascorbate, a potent reducing and hydroxylating molecule, is destroyed in these reactions. Accordingly, in the effort to control the damage done by the genetic defect the level of ascorbate is decreased, exacerbating the general deleterious effects of ascorbate deficiency.
One of the genetic defects where the ascorbate depleting steps are well characterized is the LDL receptor defect. All the expressions of LDL receptors (19) the inhibition of 3-hydroxy-3-methyl-glutaryl coenzyme A reductase in the synthesis of cholesterol (20), the protection of LDL against oxidative modification (21) and the stimulation of 7a -hydroxylase in the catabolism of cholesterol to bile acids (22). We suggest that it is ascorbate deficiency that is the real cause of the premature CVD associated with this inherited disease, exacerbated by the genetic defect.
In this context the recent study in familial hypercholesterolemic patients by Seed et al. (11) is of interest. In this study elevated LDL or the underlying genetic defect of the LDL-receptor were not correlated with CVD. Thus this genetic defect leading to ascorbate deficiency in combination with the genetic deposition for high Lp(a) levels significantly increased the risk of premature CVD.
As do genetic defects, exogenous risk factors for CVD lead to ascorbate depletion. The observed correlation between a high fat diet or cigarette smoking and CVD can also be explained as the result of induced ascorbate deficiency, caused by destruction of ascorbate in the catabolism of lipids and the effort to detoxify the substances in the smoke. With insufficient dietary ascorbate resupplementation, both endogenous and exogenous risk factors for CVD aggravate ascorbate deficiency and accelerate CVD development.
Ascorbate Deficiency and the Vascular Wall
Ascorbemia, the total depletion of ascorbate in scurvy, leads to a complete loss of the integrity and stability of the vascular wall and to the extravasation of blood into the perivascular area. Hypoascorbemia, leads to early forms of this impairment.
The vascular endothelium is directly affected by ascorbate deficiency. Characteristic features are changes in the cellular morphology and the presence of large intercellular gaps. These changes lead to the loss of the function of the endothelium as a barrier between the blood and the vascular wall, to increased permeability, and consequently to increased infiltration of plasma constituents into the vascular wall.
The extracellular matrix of the wall is affected. Collagen and elastin, the principal macromolecules of this matrix, are made from their precursors, procollagen, and proelastin, by hydroxylation of prolyl and lysyl residues. Ascorbate deficiency leads to an incomplete hydroxylation and thus weakens the extracellular matrix. Alterations of the endothelium and loose connective tissue are known to be characteristic features of atherosclerotic plaques.
To limit the fatal consequences of prolonged ascorbate deficiency metabolic counter measures were developed under strong evolutionary pressure.
Ascorbate Deficiency and Metabolic Countermeasurs
To limit the consequences of prolonged ascorbate deficiency metabolic countermeasures were developed under strong evolutionary pressure. The most detrimental effect of ascorbate depletion is blood loss. Thus ascorbate deficiency, to prevent the extravasation of blood, triggers a whole series of metabolic reactions, with the primary aim of inducing vasoconstriction and hemostasis.
It is therefore not surprising that ascorbate deficiency induces virtually all the risk factors predisposing to atherogenesis and thrombogenesis, most of them with immediate clinical significance. In the first line of defense against the danger of perivascular bleeding increased levels of thromboxane and decreased levels of prostacyclin (23) and prostaglandin E lead to vasoconstriction and hemostasis. We have shown that prolonged ascorbate deficiency increases fibrinogen and Lp(a) plasma levels and in this situation the antifibrinolyitic properties of Lp(a) become beneficial.
We are aware that there is no one-to-one relation between ascorbate and Lp(a). Lp(a) is a rather late part in a sequence of acute-phase reactants, or risk factors induced by ascorbate deficiency. Because of its lipid deposition in the vascular wall, however, Lp(a) is particularly detrimental.
The therapeutic implications are evident: ascorbate supplementation increases the levels of prostacyclin and potentially EDRF, the endothelial derived relaxing factor. This potent vasodilative factor is identical with nitric oxide and ascorbate may preserve the active form of EDRF by inhibiting oxidation to nitrogen dioxide. Simultaneously, ascorbate decreases the levels of thromboxane, fibrinogen, and Lp(a) and thereby contributes to a fundamental improvement of the risk profile in clinical cardiology.
The Roles of Lp(a) and Fibrinogen in the Vascular Wall
In the Hamburg studies Lp(a) was found mainly deposited together with fibrinogen/fibrin (10). Moreover, Lp(a) has been shown to bind to immobilized fibrinogen/fibrin (25) and evidence for a direct binding of Lp(a) to fibrinogen/fibrin in the vascular wall was reported (9). All these observations can now be explained. In ascorbate deficiency the need for increased plasma concentrations of Lp(a) and fibrinogen, for binding of Lp(a) to fibrinogen/ fibrin in the vascular wall, and for its selective retention become evident.
The hemostatic properties of Lp(a) and fibrinogen are needed to counteract the deleterious consequences of ascorbate deficiency. Lp(a) also has functions in the containment of diseases and the repair of tissues. Free-radical-induced and plasmin-induced tissue degradation are established pathways.
We have suggested that apo(a), because of many disulfide groups that can be reduced by ascorbate to thiols, can itself function as an antioxidant (1). Moreover, we now suggest that because of its homology to plasmin Lp(a) also inhibits plasmin-induced tissue degradation. The lipid content of the Lp(a) particle simultaneously provides the substrate for cell repair. In order to exert its physiological functions Lp(a) is deposited as an intact lipoprotein particle and can be isolated from the vascular wall (8). The extracellular accumulation of Lp(a) in the vascular wall is an independent pathomechanism of human CVD which is at variance with concepts suggesting the cellular uptake and degradation of lipoproteins by scavenger cells is a prerequisite for atherogenesis (2,4).
A Theory for Human Cardiovascular Disease
We are now able to present a novel pathomechanism for human cardiovascular disease. This disease is primarily a degenerative disease caused by chronic ascorbate deficiency. The extracellular deposition of Lp(a) and fibrinogen is a defense mechanism to limit the damage done by this deficiency. Under chronic conditions this defense may, however, turn into a pathologic process leading to the continued accumulation of Lp(a) and fibrinogen/fibrin in the vascular wall. Thus Lp(a) and fibrinogen/fibrin become the hallmarks of the atherosclerotic lesion (see figure).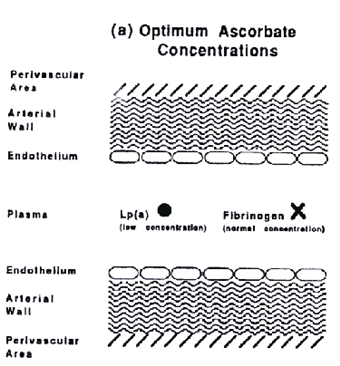
Figure a.
The impairment of the integrity of the vascular wall in ascorbate deficiency leads to increased infiltration of plasma constituents and to intimal thickening throughout the vascular system but not necessarily to the development of atherosclerotic plaques. If, however, altered hemodynamic conditions reveal the underlying impairment of the vascular wall these plaques develop.
This theory explains why human atherosclerosis develops mainly at sites of altered hemodynamic conditions such as the branching regions of coronary, cervical and cerebral arteries. It explains why the primary manifestations of human CVD is myocardial infarction and stroke, and also the increased risk of CVD associated with hypertension, where an increased systemic pressure extensively unmasks the underlying impairment of the vascular wall.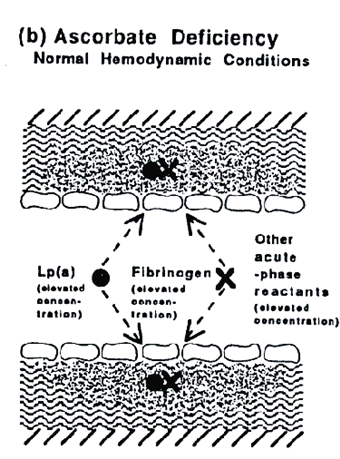
Figure b.
It is unlikely that Lp(a) primarily exerts its atherogenicity by binding to the plasminogen receptor on endothelial cells (27). These receptors are present throughout the vascular system so that such a pathomechanism would lead to increased incidence of peripheral vascular diseases and venous thrombi, which are not necessarily associated with elevated Lp(a) plasma levels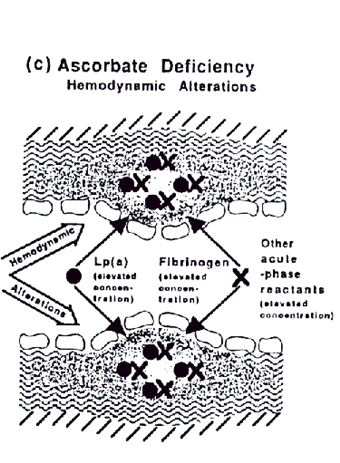
Figure c.
Peripheral Forms of Atherosclerosis
We are now able to account for another phenomenon associated with human CVD: The principle difference in the pathomechanisms leading on the one hand to atherosclerosis at predisposition sites and on the other hand to peripheral vascular disease (PVD). Myocardial infarction and stroke are by far the most frequent manifestations of CVD. The localized development of atherosclerotic plaques in these patients can only be explained if the instability of the vascular wall is the main risk factor. Elevated concentrations of plasma risk factors, e.g., cholesterol or LDL, can not explain the phenomenon of localized manifestation of CVD. They may, however, play an aggravating role in the development of CVD in the individual.
In the development of PVD, however, these plasma risk factors play a much more prominent role, exerting a direct or indirect noxious effect on the vascular wall. Consequently, this leads to atherosclerosis in the vascular periphery where the contact between noxious plasma constituents and the endothelium is prolonged. Triglyceride-rich lipoproteins, because of their enhanced susceptibility to peroxidation, are such potential challengers, leading to vascular damage in the periphery.
This theory explains the peripheral form of CVD associated with Type-III hyperlipidemia, a metabolic disorder in which triglyceride-rich lipoproteins accumulate in the plasma as very low-density lipoproteins (VLDL) and intermediate-density lipoproteins (IDL). These conditions are also characterized by a further pathomechanism of lipid deposition in the vascular wall. In addition to the extracellular deposition of Lp(a) described above, the cellular uptake of oxidatively modified lipoproteins by scavenger cells plays a more prominent role. This can also explain why foam cells are found much more frequently in the vascular wall of patients with these metabolic disorders.
A similar pathomechanism is involved in PVD associated with cigarette smoking, Oxygen free radicals from the cigarette smoke damage the endothelium directly or via oxidative modification of lipoproteins. It is noteworthy that ascorbate, the strongest antioxidant normally present in the human body, is also a potent inhibitor of these pathomechanisms.
In general, inherited metabolic disorders resulting in an elevated concentration of potentially noxious plasma constituents are frequently associated with PVD, e.g., in homocystinuria.
Of particular interest is the pathogenesis of PVD in diabetes mellitus. The glucose and ascorbate molecules share structural similarities and compete for the same transport system for cellular uptake. Elevated glucose levels competitively inhibit an optimum tissue uptake of ascorbate, leading also to a chronic ascorbate depletion of the vascular wall and its impairment. Therefore, dietary supplementation of ascorbate should lead to an effective control of diabetic angiopathy.
The different pathomechanisms leading on the one hand to CVD at predisposition sites and on the other hand to PVD are frequently interrelated. Nevertheless, their discrimination described here may prove helpful for future therapeutic approaches. Independent of the different pathomechanisms involved, ascorbate deficiency is a common denominator of human CVD.
Prophylactic and Therapeutic Considerations
The theory presented in this paper immediately suggests effective prophylactic and therapeutic treatments for most individuals at risk CVD and for CVD patients.
Prophylaxis.
Ascorbate, a potent reducing and hydroxylating agent has been shown to be effective in achieving critical prophylactic aims: lowering the plasma Lp(a) level, preventing Lp(a) deposition in the vascular wall (16), decreasing elevated LDL levels (28), increasing HDL levels (29), preventing oxidative modification of lipoproteins, protecting against oxidative damage by scavenging oxygen free radicals and by regenerating tocopherol, [ppteventing the oxidative modification of lipoproteins (30), and, above all, preserving the integrity of the vascular wall and preventing the formation of atherosclerotic plaques (16).
Moreover ascorbate hits all these therapeutic targets at the same time. It will be hard for any pharmaceutical product to surpass ascorbate, a substance that has been developed and improved by nature over billions of years. Premature atherosclerosis is essentially unknown in most animals, whereas millions of humans, with chronic ascorbate deficiency, die of atherosclerosis and related diseases each year.
Therapeusis.
Ascorbate is able not only to prevent the formation of atherosclerotic lesion but also to reduce existing plaques. It is well-established that ascorbate increases HDL plasma levels, thereby promoting reverse cholesterol transport by uptake of intra- and extracellular lipid from the vascular wall.
On the basis of our finding that plaque development is paralleled by the extracellular deposition of Lp(a) it is evident that a major focus of therapeutic development is the release of Lp(a) or its lipid component from the arterial wall. Ascorbate may be involved in two ways: by dissociating apo(a) from the LDL-like component of Lp(a), thus enhancing the lipoprotein efflux from the vascular wall and by converting lysyl residues in this wall into hydroxylysyl residues, thereby decreasing the binding affinity to components of the vascular wall by way of the lysyl haptenic group.
The efficiency of releasing Lp(a) from its bonds to fibrinogen/fibrin in the vascular wall may be considerably enhanced by administration also of small prophylactic doses of one or more inhibitors that compete with the lysyl haptenic groups [lysine, 6-aminohexanoic acid, p-aminomethylbenzoic acid, trans-4-aminomethylcyclohexane carboxylic acid, and others].
For patients with advanced cardiovascular disease therapeutic amounts of these inhibitors, together with ascorbate and as adjuncts to appropriate conventional therapy, might be prescribed, once their therapeutic effect has been clinically proved.
It might be argued that this class of substances, which are generally used as anti-fibrinolytic agents, might induce coagulative complications. These substances are, however, protease inhibitors and inhibit activation of fibrinolysis as well as the activation of coagulation (31). These substances have been used in long-term studies for different indications without compromising side effects. We have, however, not found any earlier recommendation of the use of these substances in the pharmacological treatment of cardiovascular disease. The combination of these substances with ascorbate may be considered ideal since ascorbate reduces the need for further Lp(a) deposition in the vascular wall and the inhibitors would enhance the release of already deposited Lp(a). Moreover, ascorbate is known to have anti-coagulative (32) and profibrinolytic properties.
Conclusion
The concept presented here offers for the first time a conclusive explanation for the unique features of human CVD. It can answer the questions that have remained yet unexplained by presently available hypotheses on the development of CVD (1,2,3) Ascorbate deficiency is a precondition as well as a common denominator of CVD. With rare exceptions CVD is a degenerative disease. Its leading risk factor is the instability of the vascular wall rather than any plasma constituents, and its primary pathomechanism is the deposition of Lp(a) and fibrinogen/fibrin in the vascular wall.
We can now explain why the strongest downward trend in CVD mortality of all industrialized countries occurred in the USA, the country with the highest vitamin C consumption. Moreover, we now understand why these two developments exactly parallel each other. On the basis of the scientific concept presented in this publication it is now possible to achieve a similar success also in other countries.
The pathomechanisms described here and the therapeutic conclusions presented are the solution to the puzzle of human cardiovascular disease.
We have discussed the following points in detail:
- the cause of today's most important disease by ascorbate deficiency, the result of a genetic defect in combination with inadequate intake of supplementary ascorbate;
- the regulation of plasma Lp(a) levels by ascorbate and the reasons why Lp(a) and ascorbate are found alternatively in most animal species;
- the identification of ascorbate deficiency as a common denominator of endogenous and exogenous risk factors for CVD;
- the conditions under which a physiological defense mechanism designed by nature to limit the deleterious effects of ascorbate deficiency can turn into a pathological process;
- the extracellular deposition of Lp(a) and fibrinogen/fibrin as the primary mechanism of human atherogenesis;
- the details of a comprehensive theory of human cardiovascular disease; and the difference between atherosclerosis at predisposition sites and peripheral vascular disease;
- finally, we presented prophylactic and therapeutic recommendations made on the basis of these discoveries, which may lead to a breakthrough for the prevention and treatment of human CVD.
50 years ago ascorbate deficiency was established as a prominent risk factor in CVD (33), and 37 years ago ascorbate was shown in preliminary angiographic studies to reduce atherosclerotic plaques in man (34). There is no rational explanation why these early observations of the therapeutic value of ascorbate were ignored and did not become common knowledge in the medical profession long ago.
Our publications have initiated further clinical trials. The evidence of the beneficial effects of ascorbate available now is already convincing but comprehensive clinical confirmation should soon end the decades of reluctance and skepticism. We are convinced that before long ascorbate will become the treatment of first choice for cardiovascular disease.
The therapeutic significance of our discovery is not limited to CVD; Lp(a) and ascorbate are involved in cancer, inflammatory disease, and other diseases, including the process of aging. The deposition of Lp(a) in the vicinity of disease can be conceived as a defense mechanism to contain the progression of disease, particularly at low ascorbate concentrations. The Lp(a)-ascorbate connection is a regulatory principle of nature that directly affects human health. Abolition of ascorbate deficiency may profoundly improve human health and increase life expectancy of human beings.
References
Rath M & Pauling L (1990): Proceedings of the National Academy of Sciences USA 87, 6204-6207.
Brown MS & Goldstein JL (1984): Scientific American 251, 58-66.
Ross R (1986): New England Journal of Medicine 314, 488-500.
Steinberg D, Parthasarathy S, Carew TE, Khoo JC, & Witztum JL (1989): New England Journal of Medicine 320, 915-924.
Berg K (1963): Acta Pathologica 59, 369-382.
McLean JW, Tomlinson JE, Kuang WJ, Eaton DL, Chen EY, Fless GM, Scanu AM & Lawn RM (1987): Nature 300, 132-137.
Brown MS & Goldstein JL (1987): Nature (London) 330, 113-114.
Rath M, Niendorf A, Reblin T, Dietel M, Krebber HJ & Beisiegel U (1989): Arteriosclerosis 9, 579-592.
Niendorf A, Rath M, Wolf K, Peters S, Arps H, Beisiegel U & Dietel M (1990): Virchows Archiv. A. Pathol. Anat. 417, 105-111.
Beisiegel U, Niendorf A, Wolf K, Reblin T & Rath M (1990): European Heart Journal 11, Suppl. E., 174-183.
Seed BM, Hoppichler F, Reaveley D, McCarhty S, Thompson GR, Boerwinkle E & Utermann G (1990): New England Journal of Medicine 322, 1494-1499.
Dahlen GH, Guyton JR, Attar M, Farmer JA, Kautz JA & Gotto AM Jr. (1986): Circulation 74, 758-765.
Zenker G, Koltringer P, Bone G, Kiederkorn K, Pfeiffer K & Jurgens G (1986): Stroke 17, 942-945.
Hoff HF & Gaubatz JW (1982): Atherosclerosis 42: 273-297.
Gavish D & Breslow JL (1991): Lancet 337, 203-204.
Rath M & Pauling L (1990): Proceedings of the National Academy of Sciences USA 87, 9388-9390.
Carlson LA, Hamsten A & Asplund A (1989): Journal of Internal Medicine 226, 271-276.
Pauling L (1968): Science 160, 265-271.
Aulinskas TH, Van der Westerhuyzen DR & Coetzee GA (1983): Atherosclerosis 47, 159-171.
Harwood HJ Jr, Greene YJ & Stacpoole PW (1986): Journal of Biological Chemistry 261, 7127-7135.
Frei B, England L & Ames BN (1989): Proceedings of the National Academy of Sciences USA 86, 6377-6381.
Ginter E (1973): Science 179, 702-704.
Beetens J, Coene M-C, Verheyen A, Zonnekyn L & Herman AG (1986): Prostaglandins 32, 335-352.
Loscalzo J, Weinfeld M, Fless GM & Scanu AM (1990): Arteriosclerosis 10, 240-245.
Harpel PC, Gordon BR & Parker TS (1989): Proceedings of the National Academy of Sciences USA 86, 3847-3851.
Smith EB & Cochran S (1990): Atherosclerosis 84, 173-181.
Miles LA, Fless GM, Levin EG, Scanu AM & Plow EF (1989): Nature 339, 301-303.
Ginter E (1979): Wld. Rev. Nutr. Diet. 33, 104-141.
Bates CJ, Mandal AR, Cole TJ (1977): Lancet 3, 611.
Jialal I, Vega GL & Grundy SM (1990): Atherosclerosis 82, 185-191.
Aoki N, Naito K & Yoshida N (1978): Blood 1, 1-12.
Bordia A & Verma SD (1985): Clinical Cardiology 8, 552-554.
Paterson JC (1941): Canadian Medical Association Journal 44, 114-120.
Willis GC, Light AW & Gow WQS (1954): Canadian Medical Association Journal 71, 562-568.