M. Rath, L. Pauling
Journal of Orthomolecular Medicine 1992, 7: 17-23
Summary
Most human diseases, independent of their individual genetic or exogenous origin, proliferate via similar pathomechanisms. One of these universal pathways is propagated by oxygen free radicals. Here we present another universal pathomechanism: the degradation of the connective tissue by the protease plasmin. This mechanism had been described for some diseases but its universal character has still been insufficiently understood. We propose now that the proliferation of cancer, cardiovascular disease (CVD), and also inflammatory and many other diseases depends to a varying degree on this pathomechanism.
Activated macrophages, but also cancer cells, virally transformed cells, and other pathogenic cells secrete considerable amounts of plasminogen activators, which lead to an activation of plasminogen to the protease plasmin which activates procollagenase to collagenase. The resulting degradation of the extracellular matrix is a precondition for the proliferation and the clinical manifestation of any disease. Most acute and chronic diseases make use of this pathomechanism. This pathomechanism is the exacerbation of a mechanism used under physiological conditions by a variety of cellular systems of the human body. The exacerbation under pathological conditions is the result of a chronic imbalance between activators and inhibitors of this pathway. Apoprotein (a), apo(a), by virtue of its homology to plasminogen is proposed to be a competitive endogenous inhibitor of plasmin induced proteolysis and tissue degradation. The essential amino acid L-lysine functions as an exogenous inhibitor of this pathway. Therapeutic administration of L-lysine and synthetic lysine analogs, such as tranexamic acid, should lead to an effective control of plasmin- induced tissue degradation. Comprehensive clinical confirmation of this work will particularly improve the therapeutic options for advanced forms of CVD, cancer, and inflammatory and infectious diseases, including AIDS.
Full Study
Introduction
In recent years the international research community became fascinated by a unique protein in the human body: apoprotein(a) [apo(a)]. In the three decades since its discovery apo(a) has been primarily discussed in relation to its deleterious effects on human health, in particular on cardiovascular disease (CVD). We did not accept that apo(a) should have only disadvantageous properties. According to the laws of evolution apo(a) must have beneficial properties that by far outreach its disadvantages. Consequently, we discovered that under physiological conditions apo(a) functions as an adhesive protein, mediating organ differentiation and growth. Under pathophysiological conditions apo(a) primarily substitutes for ascorbate deficiency and increases tissue stability by compensating for impaired collagen metabolism, and by promoting tissue repair (1). Moreover, we proposed that apo(a) functions as an inhibitor of important pathomechanisms involved in the proliferation of many diseases. These pathomechanisms are favored during ascorbate deficiency. One of these universal pathomechanisms is the damaging effect of oxygen free radicals, which is attenuated by the antioxidative function of apo(a) as a proteinthiol (2).
Apo(a) also led us to determine the universal importance of another pathomechanism: the enzymatic degradation of the connective tissue by the protease plasmin. We recently proposed that apo(a), by virtue of its homology to plasminogen, functions as a competitive inhibitor of plasmin- induced proteolysis (3). In this publication we describe the universal character of this mechanism and the role of apo(a) in more detail. Plasmin-induced proteolysis had been described as a pathomechanism for some diseases, e.g. cancer and certain viral diseases (4,5). In cardiovascular disease, however, this mechanism has received little, if any, attention. The insufficient understanding of the universal character of this pathomechanism is further underlined by the absence of a broad therapeutic use of L-lysine and its synthetic analogs, which are exogenous inhibitors of this pathway. The lack of this knowledge continues to have detrimental consequences for human health and it prevents millions of patients from receiving optimum treatment. It is the aim of this publication to close this gap and to provide the rationale for a broad introduction of lysine and its synthetic analogs into clinical therapy.
Plasmin-Induced Proteolysis Under Physiological Conditions
Plasmin-induced proteolysis is a physiological mechanism that occurs ubiquitously in the human body. The main cellular defense systems, monocytes, macrophages, and neutrophiles, use this mechanism for their migration through the body compartments. They secrete plasminogen activators, which then activate plasminogen to plasmin. This mechanism makes efficient use of high blood and tissue concentrations of the proenzyme, plasminogen, which represents a huge reservoir of potential proteolytic activity. The activated protease plasmin then converts procollagenases into collagenases (6), and quite possibly also activates other enzymes, leading to a local degradation of the connective tissue. This local degradation of the connective tissue paves the way for the migration of macrophages through the body. The proteolytic effect of plasmin is also involved in increasing vascular permeability (7). This effect facilitates the infiltration of monocytes and other blood cells from the circulation to the tissue sites of increased requirement. Physiological conditions in which plasmin-induced proteolysis occurs include different forms of tissue formation and reorganization such as neurogenesis, vascularization, and, quite probably, growth.
Of particular importance is plasmin-induced proteolysis during the remodulation of female reproductive organs. Under hormonal stimulation mammary and uterine cells secrete plasminogen activator and thereby initiate the morphologic changes of the organ during pregnancy and lactation (4). A particularly striking example for the effectiveness of this mechanism is ovulation. Luteinizing hormone (LH) and follicle cell stimulating hormone (FSH) stimulate the secretion of plasminogen activators from granulosa cells (8). The subsequent degradation of the ovarian connective tissue is a precondition for ovulation (Figure 1a). Similarly trophoblast cells use plasmin-induced proteolysis to invade the wall of the uterus during embryo implantation in early pregnancy. In all these conditions enzyme production is transient and is precisely regulated by hormones and other control mechanisms.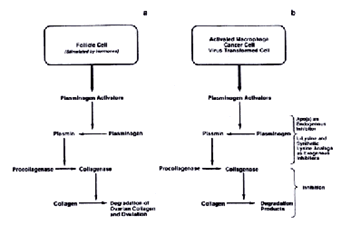
Figure 1.
Plasmin-Induced Proteolysis Under Physiological Conditions
Plasmin-induced tissue degradation contributes to the proliferation of most diseases. Of particular interest is the fact that similar mechanisms are induced by attacking pathogens as they are used by the defending host cells, e.g. macrophages. In many pathological conditions macrophages become 'activated'. This activation reflects a particular state of alert that is characterized by an abundant release of secretory products. These products include oxygen metabolites, collagenases, elastases, and a significantly increased secretion of plasminogen activators. It is immediately obvious that this mechanism needs to be precisely controlled. Therefore macrophages also secrete inhibitory products including plasmin inhibitors and a2-macroglobulin which are able to inactivate plasmin and many other proteases. Any imbalance in this control system leads to an exacerbation of this mechanism and to continued tissue degradation. Chronic activation of macrophages and an exertion of the control mechanisms eventually lead to a sustained degradation of the connective tissue and to an accelerated proliferation of the disease. It is, therefore, not unreasonable for us to propose that plasmin-induced tissue degradation contributes, to a varying degree, to the proliferation of all diseases.
This mechanism is, however, not limited to macrophages and other defense cells of the human body. In the following sections we shall discuss this pathomechanism for the most important diseases in more detail.
Cancer.
Malignant transformation of many cells of the human body leads to an uncontrolled secretion of plasminogen activators. In this situation the secretion of plasminogen activators is not a temporary event, but is rather a characteristic feature of malignant cells. The magnitude of increase in plasminogen-activator production, between 10 and 100 fold, renders this enzyme unique among the biochemical changes associated with oncogenic transformation. Moreover, plasminogen-activator secretion occurs independently of the induction mechanism and can be found as the result of oncogenic viruses or chemical carcinogens. Most importantly, the amount of plasminogen activators secreted was, in general, associated with the degree of malignancy (4,5). Immunohistological studies showed that the concentration of plasminogen activators in the vicinity of a tumor is highest at the sites of its invasive growth (9).
Because of the prominent role of plasmin-induced proteolysis in female reproductive organs under physiological conditions it is no surprise that the exacerbation of this mechanism is particularly frequent in malignancies of the female reproductive organs. Cancer cells of the breast, the uterus, the ovaries, and other organs continuously secrete increased amounts of plasminogen activators, destroy the surrounding extracellular matrix, and thereby pave the way for infiltrative growth. These mechanisms are also involved in the proliferation of prostatic cancer, one of the most frequent forms of cancer in males.
Plasmin-induced proteolysis is also critical for the metastatic spread of cancer. As discussed above, plasmin induces increased permeability of the blood vessels and thereby facilitates the systemic dissemination of tumor cells. This pathomechanism is, of course, not limited to reproductive organs. Plasmin-induced tissue degradation has been reported for tumors of the ovaries, endometrium, cervix, breast, colon, lung, skin (melanoma, and many others (4), suggesting that most cancers make use of this mechanism for their proliferation.
Infectious and inflammatory diseases.
As for transformed cells in malignancies, virally transformed cells were also found to secrete plasminogen activators (4,5). These cells activate plasminogen in their vicinity, e.g., the lung tissue, and thereby facilitate the local spread of the infection. Simultaneously, plasmin increases the permeability of the local blood vessels and thereby promotes the systemic spread of the infection.
It is not unreasonable for us to propose that other pathogens may also make use of this mechanism during the process of infection. Plasminogen activators play an important role during inflammation in general. Production of plasminogen activators by macrophages and granulocytes is closely correlated to different modulators of inflammation. Secretion of the enzyme is stimulated by asbestos, lymphokines, and interferon and is inhibited by anti-inflammatory agents such as glucocorticoids. Plasmin-induced proteolysis has been described for patients with a variety of inflammatory diseases, including chronic rheumatoid arthritis, allergic vasculitis, chronic inflamatory bowel disease, chronic sinusitis, demyelinating disease, and many others (4). Plasmin-induced tissue degradation is therefore likely to be an important pathomechanism in chronic inflammatory diseases.
Cardiovascular disease.
Activated macrophages play an important role in the pathogenesis of cardiovascular disease. Blood monocytes enter the vascular wall, where they become macrophages. Their activation inside the vascular wall is enhanced by oxidatively modified lipoproteins and other challenging mechanisms (3,10). Once they are activated a similar cascade of events occurs, as in any other disease: increased secretion of plasminogen activators, activation of procollagenases by the protease plasmin, and degradation of the connective tissue in the vascular wall. Simultaneously, plasmin increases the permeability of the vascular wall, leading to a further increase in the infiltration of plasma constituents. The perpetuation of these pathomechanisms leads to the development of atherosclerotic lesions. This mechanism is particularly effective when the vascular wall is already destabilized by a deficiency in ascorbate. As described recently in detail (3), this instability is primarily unmasked at sites of altered hemodynamic conditions, such as the branching regions of the coronary arteries. It is therefore no surprise that increased amounts of plasminogen activators were detected in these branching regions of human arteries. Moreover, atherosclerotic lesions in general were found to contain significantly higher amounts of plasminogen activators than grossly normal arterial wall (11). It is a remarkable fact that these early observations have not been followed up systematically. This negligence suggests that the universal character of uncontrolled plasmin-induced proteolysis for disease proliferation has not yet been fully understood. It is the aim of this paper to close this gap.
Apoprotein(a) - An Inhibitor of Plasmin-Induced Proteolysis
In identifying the universal importance of plasmin-induced proteolysis for most diseases we were once again guided by apo(a) and its increased demand as reflected by the elevated plasma concentrations in many pathological conditions. As discussed above, apo(a) exerts a multitude of functions under physiological and pathophysiological conditions. Here we focus on the role of apo(a) as an endogenous competitive inhibitor of plasmin-induced proteolysis and tissue degradation.
Apo(a) is a glycoprotein with a unique structure. It is essentially composed of a repetitive sequence of the kringle structures highly homologous to the kringle IV of the plasminogen molecule. The gene for apo(a) is located in the direct vicinity of the plasminogen gene on chromosome 6. It has been proposed that the apo(a) molecule derives from the plasminogen molecule or that the two genes share a common ancestral gene (12). As of today no explanation has been offered as to why among all five kringles of plasminogen it is almost exclusively kringle IV that was chosen by nature to compose the apo(a) molecule.We do not accept this selective advantage of kringle IV as a coincidence. We propose that at least one of the reasons for the repetition of kringle IV in apo(a) is closely related to the structure/function of kringle IV in the plasminogen molecule.
It is not unreasonable for us to propose that apo(a), by virtue of its multiple kringle IV structures, is a competitive inhibitor of plasmin-induced proteolysis. Apo(a) could be involved in the control of this pathway without interfering with critical functions of plasminogen mediated by other kringles of the plasminogen molecule. Consequently, the more kringle IV repeats one apo(a) molecule contains, the more effective this apo(a) isoform would be as an inhibitor. This concept could not only explain the selective advantage of kringle IV versus the other kringle structures, but it could also explain the great variation in genetically determined plasma Lp(a) concentrations, which largely reflect the inverse relation between the number of intramolecular kringle IV repeats and the synthesis rate of apo(a) molecules.
Supportive evidence for a role of apo(a) in the control of plasmin- induced proteolysis is also provided by a number of observations. Apo(a) has been shown to attenuate tissue-plasminogen-activator-induced fibrinolysis and competitively interfere with plasminogen- and plasmin- induced pathways (review in 14). Moreover, immunohistological studies in various diseases showed a preferential deposition of apo(a) at the site of increased demand for a control of plasmin-induced proteolysis. In several hundred vascular specimens representing various degrees of cardiovascular disease apo(a) was found primarily to be located in the subendothelium, quite possibly counteracting the increased endothelial permeability. In advanced atherosclerotic lesions apo(a) was preferentially found around the lesion core, particularly at the edges of the lesion (15), the main sites of chronic repair processes. In a comprehensive morphological study in different forms of cancer apo(a) was found to be deposited in the vicinity of the cancer process (Dr. A. Niendorf, personal communication). Both studies were conducted with the same monoclonal antibodies not cross-reacting with plasminogen. A preliminary report is also available for the deposition of apo(a) in the microvasculature of inflammatory processes (16). We predict that apo(a) will also be found to play an important role in the containment of infectious diseases, including AIDS. The role of apo(a) as a competitive inhibitor of plasmin-induced proteolysis is not limited to pathological conditions. An increased demand of apo(a) was also observed during the period of uterus transformation in early pregnancy (17).
In summary, apo(a) is suggested to be an important element in the endogenous control system of plasmin-induced proteolysis. Apo(a) may back-up antiplasmin and other endogenous inhibitors of this pathway particularly during chronic activation of this mechanism. Beside endogenous inhibitors of plasmin-induced tissue degradation there are also exogenous inhibitors. The universal importance of the pathomechanism described here immediately suggests the great value of these exogenous inhibitors in the therapy of many diseases.
The Therapeutic Use of Lysine and Synthetic Lysine Analogs
Lysine, an essential amino acid, is the most important naturally- occurring inhibitor of this pathway. As opposed to the competitive inhibition by apo(a), lysine inhibits plasmin-induced proteolysis in a direct way. Lysine attenuates an overshooting activation of plasmin, at least in part, by occupying the lysine binding sites in the plasminogen molecule. Since lysine is an essential amino acid, its availability is not regulated endogenously. Insufficient dietary lysine intake invariably leads to a deficiency of this amino acid and thereby weakens the natural defense against this pathomechanism. Moreover, chronic activation of plasminogen by cancer cells, virally transformed cells, or macrophages leads to an additional relative lysine deficiency and thereby to an acceleration of the underlying disease. The therapeutic value of lysine has been documented for a variety of diseases, including viral diseases (18), and recently in combination with ascorbate for cardiovascular disease (19).
Synthetic lysine analogs such as epsilon-aminocaproic acid, para-aminomethylbenzoic acid and trans-aminocyclohexanoic acid (tranexamic acid) are potent inhibitors of plasmin-induced proteolysis. These substances, in particular tranexamic acid, have been successfully used in the treatment of a variety of pathological conditions, such as angiohematoma, colitis ulcerosa, and others. Most remarkable results were reported from the treatment of patients with late-stage cancer of the breast (20) and the ovaries (21) and also for cancer of other origins (22). We have recently suggested the therapeutic use of synthetic lysine analogs for the reduction of atherosclerotic plaques (3).
On the basis of the work presented here, comprehensive clinical studies should be initiated to establish the critical role of lysine in the prevention and treatment of various diseases without delay. A daily intake of 5 grams of lysine and more (19,23) has been described to be without side effects. On the basis of the encouraging therapeutic results with tranexamic acid, particularly in inhibiting and reducing late-stage cancer, these substances should now be extensively tested for a broad introduction into clinical therapy, particularly for advanced forms of cancer, CVD, and AIDS. A possible explanation of why this has not happened long ago may be the argument that these substances may induce coagulative complications. They are , however, protease inhibitors and inhibit not only fibrinolysis but also coagulation (24). Moreover, tranexamic acid has been given for more than 10 years without clinical complications (25). We have proposed that the risk of any hemostatic complication will be further reduced by a combination of these compounds with ascorbate and other vitamins with anticoagulative properties (3). This medical consideration is, however, not the only factor why these compounds are not used much more frequently and why thousands of patients are still deprived of optimum therapy. There is also an economic factor. Patent protection is a guiding principle of any pharmaceutical company in developing or marketing a drug. Lysine, like many other nutrients, is not patentable and the patents for the clinically approved synthetic lysine analogs, including tranexamic acid, have expired. The negligence of these substances may be explainable from the economic point of view; from the perspective of human health there is no justification for this delay.
Conclusion
Here we have described plasmin-induced proteolysis as a universal pathomechanism propagating cancer, and cardiovascular, inflammatory, and many other diseases. Plasmin-induced tissue degradation under pathological conditions is an exacerbation of a physiological mechanism. Apo(a) is suggested to function as a competitive endogenous inhibitor of this pathway. On the basis of the selective advantage of apo(a) in the evolution of man it comes as no surprise that apo(a) should lead us on the way to recognize the universal importance of this pathomechanisms. Further clinical confirmation of the therapeutic value of lysine and its synthetic analogs may provide new options for an effective therapy for millions of people. We predict that the use of lysine and synthetic lysine analogs, particularly in combination with ascorbate, will lead to a breakthrough in the control of many forms of cancer and infectious diseases, including AIDS, as well as many other diseases.
Acknowledgements
We thank Dr. Alexandra Niedzwiecki for helpful discussions, Rosemary Babcock for library services, Jolanta Walechiewicz for graphical assistance, Martha Best and Dorothy Munro for secretarial help.
References
1. Rath M, Pauling L. Apoprotein(a) is an adhesive protein. J. Orthomolecular Med.1991;6:139-143.
2. Rath M, Pauling L. Hypothesis: Lipiprotein(a) is a surrogate for ascorbate. Proc.Natl.Acad.Sci.USA 1990; 87:6204-6207.
3. Rath M, Pauling L. Solution of the puzzle of human cardiovascular disease: Its primary cause is ascorbate deficiency, leading to the deposition of lipoprotein(a) and fibrinogen/fibrin in the vascular wall. J. Orthomolecular Med.1991;6:125-134.
4. Dan¿ K, Andreasen PA, Gr¿ndahl-Hansen J, Kristensen P, Nielsen LS and Skriver L: Plasminogen activators, tissue degradation, and cancer. Advances in Cancer Research 1985; Vol 44, Academic Press, .
5. Reich E: Activation of plasminogen: a general mechanism for producing localized extracellular proteolysis. Molecular Basis of Biological Degradative Processes. Berlin RD, Herrmann H, Lepow TH, Tanzov T (eds), 1978, Academic Press Inc.,New York .
6. Werb Z, Mainardi CL, Vater CA, and Harris Jr ED: Endogenous activation of latent collagenase by rheumatoid synovial cells. N.Engl.J.Med.1977 #18; 296:
7. Ratnoff OD. Increased vascular permeability induced by human plasmin. In: Vascular Permeability and Plasmin. 1965.
8. Strickland S & Beers WH. Studies on the role of plasminogen activator in ovulation. J.Biol.Chem.1976; 251:5694-5702.
9. Skriver L, Larsson L-I, Kielberg V, Nielsen LS, Andresen PB, Kristensen P, & Dan¿ K. Immunocytochemical localization of urokinase-type plasminogen activator in Lewis lung carcinoma. J.Cell Biol. 1984; 99:752-757.
10. Steinberg D, Parthasarathy S, Carew TE, & Witztum JL. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N. Engl. J. Med. 1989; 320:915-924.
11. Smokovitis A: A new hypothesis: possible mechanisms in the involvement of the increased plasminogen activator activity in branching regions of the aorta in the initiation of atherosclerosis. Thromb-Haemost. 1980; 43(2):141-148.
12. McLean JW, Tomlinson JE, Kuang W-J, Eaton DL, Chen EY, Fless GM, Scanu AM, and Lawn RM. cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature 1987;330:132-137.
13. Trexler M, V‡li & Patthy L. Structure of the w-aminocarboxylic acid-binding sites of human plasminogen. J.Biol.Chem. 1982; 257:7401-7406.
14. Edelberg JM, Pizzo SV: Lipoprotein(a): The link between impaired fibrinolysis and atherosclerosis. Fibrinolysis 1991;5:135-143.
15. Niendorf A, Rath M, Wolf K, Peters S, Arps H, Beisiegel U and Dietel M: Morphological detection and quantification of lipoprotein(a) deposition in atheromatous lesions of human aorta and coronary arteries. Virchow's Archiv A Pathol. Anat. 1990;417:105-111.
16. Etingin OR, Hajjar DP, Hajjar KA, Harpel PC & Nachman RL. Lipoprotein(a) regulates plasminogen activator inhibitor-1 expression in endothelial cells. J.Biol.Chem.1991; 266:2459-2465.
17. Zechner R, Desoye G, Schweditsch MO, Pfeiffer KP & Kostner GM. Fluctuations of plasma lipoprotein-a concentrations during pregnancy and post partum. Metabolism 1986; 35:333-336.
18. Griffith RS, Walsh DE, Myrmel KH, Thompson RW, Behforooz A. Success of L-lysine therapy in frequently recurrent herpes simplex infection. Dermatologica 1987; 130:183-190.
19. Pauling L. Case report: Lysine/ascorbate-related amelioration of angina pectoris. J. Orthomolecular Med.1991;6:144-146.
20. ?stedt B, Mattsson W, TropŽ C. Treatment of advanced breast cancer with chemotherapeutics and inhibition of coagulation and fibrinolysis. Acta Med. Scand. 1977;201:491-493.
21. ?stedt B, Glifberg I, Mattsson W, TropŽ C. Arrest of growth of ovarian tumor by tranexamic acid. JAMA 1977; 238:154.
22. Markus G. The role of hemostasis and fibrinolysis in the metastatic spread of cancer. Seminars in Thrombosis and Hemostasis 1984: 10;61-70.
23. Rose WC, Johnson JE & Haines W. The amino acid requirement of man. J Biol Chem 1950;182:541-556.
24. Aoki N, Naito K, & Yoshida N. Inhibition of platelet aggregation by protease inhibitors. Possible involvement of proteases in platelet aggregation. Blood 1978; 52:1-12.
25. Munch EP & Weeke B. Non-hereditary angioedema treated with tranexamic acid. Allergy 1985; 40: 92-97.