M. Rath, L. Pauling
Journal of Orthomolecular Medicine 1992, 7: 5-15
Summary
Until now therapeutic concepts for human cardiovascular disease (CVD) were targeting individual pathomechanisms or specific risk factors. On the basis of genetic, metabolic, evolutionary, and clinical evidence we present here a unified pathogenetic and therapeutic approach. Ascorbate deficiency is the precondition and common denominator of human CVD. Ascorbate deficiency is the result of the inability of man to synthesize ascorbate endogenously in combination with insufficient dietary intake.
The invariable morphological consequences of chronic ascorbate deficiency in the vascular wall are the loosening of the connective tissue and the loss of the endothelial barrier function. Thus human CVD is a form of pre-scurvy. The multitude of pathomechanisms that lead to the clinical manifestation of CVD are primarily defense mechanisms aiming at the stabilization of the vascular wall.
After the loss of endogenous ascorbate production during the evolution of man these defense mechanisms became life-saving. They counteracted the fatal consequences of scurvy and particularly of blood loss through the scorbutic vascular wall. These countermeasures constitute a genetic and a metabolic level. The genetic level is characterized by the evolutionary advantage of inherited features that lead to a thickening of the vascular wall, including a multitude of inherited diseases. The metabolic level is characterized by the close connection of ascorbate with metabolic regulatory systems that determine the risk profile for CVD in clinical cardiology today.
The most frequent mechanism is the deposition of lipoproteins, particularly lipoprotein(a) [Lp(a)], in the vascular wall. With sustained ascorbate deficiency, the result of insufficient ascorbate uptake, these defense mechanisms overshoot and lead to the development of CVD.
Premature CVD is essentially unknown in all animal species that produce high amounts of ascorbate endogenously. In humans, unable to produce endogenous ascorbate, CVD became one of the most frequent diseases. The genetic mutation that rendered all human beings today dependent on dietary ascorbate is the universal underlying cause of CVD.
Optimum dietary ascorbate intake will correct this common genetic defect and prevent its deleterious consequences. Clinical confirmation of this theory should largely abolish CVD as a cause for mortality in this generation and future generations of mankind.
Full Study
Introduction
We have recently presented ascorbate deficiency as the primary cause of human CVD. We proposed that the most frequent pathomechanism leading to the development of atherosclerotic plaques is the deposition of Lp(a) and fibrinogen/fibrin in the ascorbate-deficient vascular wall (1, 2). In the course of this work we discovered that virtually every pathomechanism for human CVD known today can be induced by ascorbate deficiency. Beside the deposition of Lp(a) this includes such seemingly unrelated processes as foam cell formation and decreased reverse-cholesterol transfer, and also peripheral angiopathies in diabetic or homocystinuric patients. We did not accept this observation as a coincidence.
Consequently we proposed that ascorbate deficiency is the precondition as well as a common denominator of human CVD. This far-reaching conclusion deserves an explanation; it is presented in this paper. We suggest that the direct connection of ascorbate deficiency with the development of CVD is the result of extraordinary pressure during the evolution of man. After the loss of the endogenous ascorbate production in our ancestors, fatal blood-loss through the scorbutic vascular wall became a life-threatening condition. The resulting evolutionary pressure favored genetic and metabolic mechanisms predisposing to CVD.
The Loss of Endogenous Ascorbate Production in the Ancestor of Man
With few exceptions all animals synthesize their own ascorbate by conversion from glucose. In this way they manufacture a daily amount of ascorbate that varies between about 1 gram and 20 grams, when compared to the human body weight. About 40 million years ago the ancestor of man lost the ability for endogenous ascorbate production. This was the result of a mutation of the gene encoding for the enzyme L-gulono-g -lactone oxidase (GLO), a key enzyme in the conversion from glucose to ascorbate. As a result of this mutation all descendants became dependent on dietary ascorbate intake.
The precondition for the mutation of the GLO gene was a sufficient supply of dietary ascorbate. Our ancestors at that time lived in tropical regions. Their diet consisted primarily of fruits and other forms of plant nutrition that provided a daily dietary ascorbate supply in the range of several hundred milligrams to several grams per day. When our ancestors left this habitat to settle in other regions of the world the availability of dietary ascorbate dropped considerably and they became prone to scurvy.
Fatal Blood Loss Through the Scorbutic Vascular Wall - An Extraordinary Challenge to the Evolutionary Survival of Man
Scurvy is a fatal disease. It is characterized by structural and metabolic impairment of the human body, particularly by the destabilization of the connective tissue. Ascorbate is essential for an optimum production and hydroxylation of collagen and elastin, key constituents of the extracellular matrix. Ascorbate depletion thus leads to a destabilization of the connective tissue throughout the body. One of the first clinical signs of scurvy is perivascular bleeding. The explanation is obvious: Nowhere in the body does there exist a higher pressure difference than in the circulatory system, particularly across the vascular wall. The vascular system is the first site where the underlying destabilization of the connective tissue induced by ascorbate deficiency is unmasked, leading to the penetration of blood through the permeable vascular wall. The most vulnerable sites are the proximal arteries, where the systolic blood pressure is particularly high. The increasing permeability of the vascular wall in scurvy leads to petechiae and ultimately hemorrhagic blood loss.
Scurvy and scorbutic blood loss decimated the ship crews in earlier centuries within months. It is thus conceivable that during the evolution of man periods of prolonged ascorbate deficiency led to a great death toll. The mortality from scurvy must have been particularly high during the thousands of years the ice ages lasted and in other extreme conditions, when the dietary ascorbate supply approximated zero. We therefore propose that after the loss of endogenous ascorbate production in our ancestors, scurvy became one of the greatest threats to the evolutionary survival of man. By hemorrhagic blood loss through the scorbutic vascular wall our ancestors in many regions may have virtually been decimated and brought close to extinction.
The morphologic changes in the vascular wall induced by ascorbate deficiency are well characterized: the loosening of the connective tissue and the loss of the endothelial barrier function. The extraordinary pressure by fatal blood loss through the scorbutic vascular wall favored genetic and metabolic countermeasures attenuating increased vascular permeability.
Ascorbate Deficiency and Genetic Countermeasures
The genetic countermeasures are characterized by an evolutionary advantage of genetic features and include inherited disorders that are associated with atherosclerosis and CVD. With sufficient ascorbate supply these disorders stay latent. In ascorbate deficiency, however, they become unmasked, leading to an increased deposition of plasma constituents in the vascular wall and other mechanisms that thicken the vascular wall. This thickening of the vascular wall is a defense measure compensating for the impaired vascular wall that had become destabilized by ascorbate deficiency. With prolonged insufficient ascorbate intake in the diet these defense mechanisms overshoot and CVD develops.
The most frequent mechanism to counteract the increased permeability of the ascorbate-deficient vascular wall became the deposition of lipoproteins and lipids in the vessel wall. Another group of proteins that generally accumulate at sites of tissue transformation and repair are adhesive proteins such as fibronectin, fibrinogen, and particularly apo(a). It is therefore no surprise that Lp(a), a combination of the adhesive protein apo(a) with a low density lipoprotein (LDL) particle, became the most frequent genetic feature counteracting ascorbate deficiency (1). Beside lipoproteins, certain metabolic disorders, such as diabetes and homocysteinuria, are also associated with the development of CVD. Despite differences in the underlying pathomechanism, all these mechanisms share a common feature: they lead to a thickening of the vascular wall and thereby can counteract the increased permeability in ascorbate deficiency.
In addition to these genetic disorders, the evolutionary pressure from scurvy also favored certain metabolic countermeasures.
Ascorbate Deficiency and Metabolic Countermeasures
The metabolic countermeasures are characterized by the regulatory role of ascorbate for metabolic systems determining the clinical risk profile for CVD. The common aim of these metabolic regulations is to decrease the vascular permeability in ascorbate deficiency. Low ascorbate concentrations therefore induce vasoconstriction, hemostasis and affect vascular wall metabolism in favor of atherogenesis. Towards this end ascorbate interacts with lipoproteins, coagulation factors, prostaglandins, nitric oxides, and second messenger systems such as cyclic monophosphates (1, 3-5). It should be noted that ascorbate can affect these regulatory levels in a multiple way. In lipoprotein metabolism low density lipoproteins (LDL), Lp(a), and very low density lipoproteins (VLDL) are inversely correlated with ascorbate concentrations, whereas ascorbate HDL levels are positively correlated. Similarly, in prostaglandin metabolism ascorbate increases prostacyclin and prostaglandin E concentrations and decreases thromboxane levels. In general, ascorbate deficiency induces vascular constriction and hemostatis, as well as cellular and extracellular defense measures in the vascular wall.
In the following sections we will exemplify the role of ascorbate for frequent and well established pathomechanisms of human CVD. In general, the inherited disorders described below are polygenic. Their separate description, however, will allow the characterization of the role of ascorbate on the different genetic and metabolic levels.
Apo(a) and Lp(a), the Most Effective and Most Frequent Countermeasures
After the loss of endogenous ascorbate production, apo(a) and Lp(a) were greatly favored by evolution. The frequency of occurrence of elevated Lp(a) plasma levels in species that had lost the ability to synthesize ascorbate is so great that we formulated the theory that apo(a) functions as a surrogate for ascorbate (6). There are several genetically determined isoforms of apo(a). They differ in the number of kringle repeats and in their molecular size (7). An inverse relation between the molecular size of apo(a) and the number of synthesized Lp(a) molecules has been established. Patients with the high molecular weight apo(a) isoform carry fewer LDL particles in their Lp(a) fraction. Vice versa, patients with the genetic pattern of low apo(a) isoform have more LDL particles in their Lp(a) plasma fraction and thus have increased Lp(a) plasma levels. In most population studies the genetic pattern of high apo(a) isoform/low Lp(a) plasma level proved to be the most advantageous and therefore most frequent pattern.
In ascorbate deficiency Lp(a) is selectively retained in the vascular wall. Apo(a) counteracts increased permeability by compensating for collagens, by its binding to fibrin, as a proteinthiol and antioxidant, and as an inhibitor of plasmin-induced proteolysis (1). Moreover, as an adhesive protein apo(a) is effective in tissue-repair processes (8). Chronic ascorbate deficiency leads to a sustained accumulation of Lp(a) in the vascular wall. This leads to the development of atherosclerotic plaques and premature CVD particularly in individuals with genetically determined high plasma Lp(a) levels. Because of its association with apo(a), Lp(a) is the most specific repair particle among all lipoproteins. Lp(a) is predominantly deposited at predisposition sites and it is therefore found to be significantly correlated with coronary, cervical, and cerebral atherosclerosis but not with peripheral vascular disease.
The mechanism by which ascorbate resupplementation prevents CVD in any condition is by maintaining the integrity and stability of the vascular wall. In addition, ascorbate exerts in the individual a multitude of metabolic effects that prevent the exacerbation of a possible genetic predisposition and the development of CVD. If the predisposition is a genetic elevation of Lp(a) plasma levels the specific regulatory role of ascorbate is the decrease of apo(a) synthesis in the liver and thereby the decrease of Lp(a) plasma levels. Moreover, ascorbate decreases the retention of Lp(a) in the vascular wall by lowering fibrinogen synthesis and by increasing the hydroxylation of lysine residues in vascular wall constituents, thereby reducing the affinity for Lp(a) binding (1).
In about half of the CVD patients the mechanism of Lp(a) deposition contributes significantly to the development of atherosclerotic plaques. Other lipoprotein disorders are also frequently part of the polygenic pattern predisposing the individual patient to CVD in the individual.
Other Lipoprotein Disorders Associated with CVD
In a large population study Goldstein identified three frequent lipid disorders, familial hypercholesterolemia, familial hypertriglyceridemia, and familial combined hyperlipidemia (9). Ascorbate deficiency unmasks these underlying genetic defects and leads to an increased plasma concentration of lipids (e.g. cholesterol, triglycerides) and lipoproteins (e.g. LDL, VLDL) as well as to their deposition in the impaired vascular wall. As with Lp(a), this deposition is a defense measure counteracting the increased permeability. It should, however, be noted that the deposition of lipoproteins other than Lp(a) is a less specific defense mechanism and frequently follows Lp(a) deposition. Again, these mechanisms function as a defense only for a limited time. With sustained ascorbate deficiency the continued deposition of lipids and lipoproteins leads to atherosclerotic plaque development and CVD. Some mechanisms will be described in more detail:
Hypercholesterolemia, LDL-receptor defect.
A multitude of genetic defects lead to an increased synthesis and/or a decreased catabolism of cholesterol or LDL. A well characterized although rare defect is the LDL-receptor defect. Ascorbate deficiency unmasks these inherited metabolic defects and leads to an increased plasma concentration of cholesterol-rich lipoproteins, e.g. LDL, and their deposition in the vascular wall. Hypercholesterolemia increases the risk for premature CVD primarily when combined with elevated plasma levels of Lp(a) or triglycerides.
The mechanisms by which ascorbate resupplementation prevents the exacerbation of hypercholesterolemia and related CVD include an increased catabolism of cholesterol. In particular, ascorbate is known to stimulate 7a-hydroxylase, a key enzyme in the conversion of cholesterol to bile acids and to increase the expression of LDL receptors on the cell surface. Moreover, ascorbate is known to inhibit endogenous cholesterol synthesis as well as oxidative modification of LDL (1).
Hypertriglyceridemia, Type III hyperlipidemia.
A variety of genetic disorders lead to the accumulation of triglycerides in the form of chylomicron remnants, VLDL and intermediate density lipoproteins (IDL) in plasma. Ascorbate deficiency unmasks these underlying genetic defects and the continued deposition of triglyceride-rich lipoproteins in the vascular wall leads to CVD development. These triglyceride-rich lipoproteins are particularly subject to oxidative modification, cellular lipoprotein uptake, and foam cell formation. In hypertriglyceridemia non specific foam cell formation has been observed in a variety of organs (10). In the vascular wall foam cell formation, although a less specific repair mechanism than the extracellular deposition of Lp(a), may have also conferred stability on the ascorbate-deficient vascular wall.
Ascorbate resupplementation prevents the exacerbation of CVD associated with hypertriglyceridemia, Type III hyperlipidemia, and related disorders by stimulating lipoprotein lipases and thereby enabling a normal catabolism of triglyceride-rich lipoproteins (11). Ascorbate prevents the oxidative modification of these lipoproteins, their uptake by scavenger cells and foam cell formation. Moreover, we propose here that, analogous to the LDL receptor, ascorbate also increases the expression of the receptors involved in the metabolic clearance of triglyceride-rich lipoproteins, such as the chylomicron remnant receptor.
The degree of build-up of atherosclerotic plaques in patients with lipoprotein disorders is determined by the rate of deposition of lipoproteins and by the rate of the removal of deposited lipids from the vascular wall. It is therefore not surprising that ascorbate is also closely connected with this reverse pathway.
Hypoalphalipoproteinemia.
A frequent lipoprotein disorder is the genetically determined decreased synthesis of HDL particles. HDL is part of the 'reverse-cholesterol-transport' pathway and is critical for the transport of cholesterol and also other lipids from the body periphery to the liver. In ascorbate deficiency this genetic defect is unmasked resulting in decreased HDL levels and a decreased reverse transport of lipids from the vascular wall to the liver. This mechanism is highly effective and the genetic disorder hypoalphalipoproteinemia was greatly favored during evolution.
With ascorbate resupplementation HDL production increases (12), leading to an increased uptake of lipids deposited in the vascular wall and to a decrease of the atherosclerotic lesion. A look back in evolution underlines the importance of this mechanism. During the winter seasons, with low ascorbate intake, our ancestors became dependent on protecting their vascular wall by the deposition of lipoproteins and other constituents. During spring and summer seasons the ascorbate content in the diet increased significantly and mechanisms were favored that decreased the vascular deposits under the protection of increased ascorbate concentration in the vascular tissue. It is not unreasonable for us to propose that ascorbate can reduce fatty deposits in the vascular wall within a relatively short time. In an earlier clinical study it was shown that 500 mg of dietary ascorbate per day can lead to a reduction of atherosclerotic deposits within 2 to 6 months (13).
This concept, of course, also explains why heart attack and stroke occur today with a much higher frequency in winter than during spring and summer, the seasons with increased ascorbate intake.
Other Inherited Metabolic Disorders Associated with CVD
Beside lipoprotein disorders many other inherited metabolic diseases are associated with CVD. Generally these disorders lead to an increased concentration of plasma constituents that directly or indirectly damage the integrity of the vascular wall. Consequently these diseases lead to peripheral angiopathies as observed in diabetes, homocysteinuria, sickle-cell anemia (the first molecular disease described (14)), and many other genetic disorders. Similar to lipoproteins the deposition of various plasma constituents as well as proliferative thickening provided a certain stability for the ascorbate-deficient vascular wall. We illustrate this principle for diabetic and homocystinuric angiopathy.
Diabetic angiopathy.
The pathomechanism in this case involves the structural similarity between glucose and ascorbate and the competition of these two molecules for specific cell surface receptors (15,16). Elevated glucose levels prevent many cellular systems in the human body, including endothelial cells, from optimum ascorbate uptake. Ascorbate deficiency unmasks the underlying genetic disease, aggravates the imbalance between glucose and ascorbate, decreases vascular ascorbate concentration, and thereby triggers diabetic angiopathy.
Ascorbate resupplementation prevents diabetic angiopathy by optimizing the ascorbate concentration in the vascular wall and also by lowering insulin requirement (17).
Homocystinuric angiopathy.
Homocystinuria is characterized by the accumulation of homocyst(e)ine and a variety of its metabolic derivatives in the plasma, the tissue and the urine as the result of decreased homocysteine catabolism (18). Elevated plasma concentrations of homocyst(e)ine and its derivatives damage the endothelial cells throughout the arterial and venous system. Thus homocystinuria is characterized by peripheral vascular disease and thromboembolism. These clinical manifestations have been estimated to occur in 30 per cent of the patients before the age of 20 and in 60 per cent of the patients before the age of 40 (19).
Ascorbate resupplementation prevents homocystinuric angiopathy and other clinical complications of this disease by increasing the rate of homocysteine catabolism (20).
Thus, ascorbate deficiency unmasks a variety of individual genetic predispositions that lead to CVD in different ways. These genetic disorders were conserved during evolution largely because of their association with mechanisms that lead to the thickening of the vascular wall. Moreover, since ascorbate deficiency is the underlying cause of these diseases, ascorbate resupplementation is the universal therapy.
The Determining Principles of This Theory
The determining principles of this comprehensive theory are schematically summarized in Figures 1 to 3.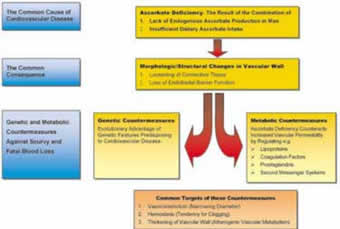
Figure 1.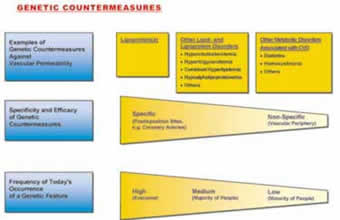
Figure 2.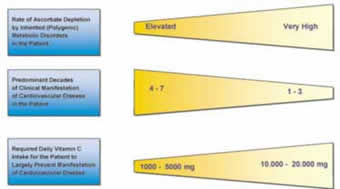
Figure 3.
1. Cardiovascular disease is the direct consequence of the inability for endogenous ascorbate production in man in combination with low dietary ascorbate intake
2. Ascorbate deficiency leads to increased permeability of the vascular wall by the loss of the endothelial barrier function and the loosening of the vascular connective tissue.
3. After the loss of endogenous ascorbate production scurvy and fatal blood loss through the scorbutic vascular wall rendered our ancestors in danger of extinction. Under this evolutionary pressure over millions of years genetic and metabolic countermeasures were favored that counteract the increased permeability of the vascular wall.
4. The level is characterized by the fact that inherited disorders associated with CVD became the most frequent among all genetic predispositions. Among those predispositions lipid and lipoprotein disorders occur particularly often.
5. The metabolic level is characterized by the direct relation between ascorbate and virtually all risk factors of clinical cardiology today. Ascorbate deficiency leads to vasoconstriction and hemostasis and affects the vascular wall metabolism in favor of atherogenesis. The genetic level can be further characterized. The more effective and specific a certain genetic feature counteracted the increasing vascular permeability in scurvy, the more advantageous it became during evolution and, generally, the more frequently this genetic feature occurs today.
7. The deposition of Lp(a) is the most effective, most specific, and therefore most frequent of these mechanisms. Lp(a) is preferentially deposited at predisposition sites. In chronic ascorbate deficiency the accumulation of Lp(a) leads to the localized development of atherosclerotic plaques and to myocardial infarction and stroke.
8. Another frequent inherited lipoprotein disorder is hypoalphalipoproteinemia. The frequency of this disorder again reflects its usefulness during evolution. The metabolic upregulation of HDL synthesis by ascorbate became an important mechanism to reverse and decrease existing lipid deposits in the vascular wall.
9. The vascular defense mechanisms associated with most genetic disorders is unspecific. These mechanisms can aggravate the development of atherosclerotic plaques at predisposition sites. Other unspecific mechanisms lead to peripheral forms of atherosclerosis by causing a thickening of the vascular wall throughout the cardiovascular system. This peripheral form of vascular disease is characteristic for angiopathies associated with Type III hyperlipidemia, diabetes, and many other inherited metabolic diseases.
10. Of particular advantage during evolution and therefore particularly frequent today are those genetic features that protect the ascorbate-deficient vascular wall until the end of the reproduction age. By favoring these disorders nature decided for the lesser of two evils: the death from CVD after the reproduction age rather than death from scurvy at a much earlier age. This also explains the rapid increase of the CVD mortality today from the 4th decade onwards.
11. After the loss of endogenous ascorbate production the genetic mutation rate in our ancestors increased significantly (21). This was an additional precondition favouring not only the advantage of apo(a) and Lp(a) but also of many other genetic countermeasures associated with CVD.
Genetic predispositions are characterized by the rate of ascorbate depletion in a multitude of metabolic reactionsspecific for the genetic disorder (22). The overall rate of ascorbate depletion in an individual is largely determined by polygenic pattern of disorders. The earlier the ascorbate reserves in the body are depleted without being resupplemented, the earlier CVD develops.
13. The genetic predispositions with the highest probability for early clinical manifestation require the highest amount of ascorbate resupplementation in the diet to prevent CVD development. The amount of ascorbate for patients at high risk should be comparable to the amount of ascorbate our ancestors synthesized in their body before they lost this ability: between 10,000 and 20,000 milligrams per day.
14. Optimum ascorbate resupplementation prevents the development of CVD independent of the individual predisposition or pathomechanism. Ascorbate reduces existing atherosclerotic deposits and thereby decreases the risk for myocardial infarction and stroke. Moreover, ascorbate can prevent blindness and organ failure in diabetic patients, thromboembolism in homocystinuric patients and many other manifestations of CVD.
Conclusion
In this paper we present a unified theory of human CVD. This disease is the direct consequence of the inability of man to synthesize ascorbate in combination with insufficient intake of ascorbate in the modern diet. Since ascorbate deficiency is the common cause of human CVD, ascorbate resupplementation is the universal treatment for this disease. The available epidemiological and clinical evidence is reasonably convincing. Further clinical confirmation of this theory should lead to the abolition of CVD as a cause of human mortality for the present generation and future generations of mankind.
References
1. Rath, M, Pauling L. Solution of the puzzle of human cardiovascular disease: Its primary cause is ascorbate deficiency, leading to the deposition of lipoprotein(a) and fibrinogen/fibrin in the vascular wall. Journal of Orthomolecular Med 1991;6:125-134.
2. Pauling L, Rath M. Vitamin C and lipoprotein(a) in relation to cardiovascular disease and other diseases. Journal of Applied Nutrition 1992; this issue.
3. Ginter E. Marginal vitamin C deficiency, lipid metabolism, and atherosclerosis. Lipid Research 1973;16:162-220.
4. Third Conference on Vitamin C, Annals of the New York Academy of Sciences 498 (BurnsJJ, Rivers JM, Machlin LJ, eds) 1987.
5. Pauling L. How to Live Longer and Feel Better 1986; Freeman, New York.
6. Rath M, Pauling L. Hypothesis: Lipoprotein(a) is a surrogate for ascorbate. Proceedings of the National Academy of Sciences USA 1990;87:6204-6207.
7. Koschinsky ML, Beisiegel U, Henne-Bruns D, Eaton DL, Lawn RM. Apolipoprotein(a) size heterogeneity is related to variable number of repeat sequences in its mRNA. Biochemistry 1990;29:640-644.<
8. Rath M, Pauling L. Apoprotein(a) is an adhesive protein. Journal of Orthomolecular Medicine 1991;6:139-143.
9. Goldstein JL, Schrott HG, Hazzard WR, Bierman EL, Motulsky AG. Hyperlipidemia in coronary heart disease. Journal of Clinical Investigation 1973; 52:1544-1568.
10. Roberts WC, Levy RI, Fredrickson DS. Hyperlipoproteinemia-A review of the five types, with first report of necropsy findings in type 3. Archives of Pathology 1970;59:46-56.
11. Sokoloff B, Hori M, Saelhof CC, Wrzolek T, Imai T. Aging, atherosclerosis and ascorbic acid metabolism. Journal of the American Geriatric Society 1966; 14:1239-1260
12. Jacques PF, Hartz SC, McGandy RB, JacobRA, Russell RM. Vitamin C and blood lipoproteins in an elderly population. Third Conference on Vitamin C, Annals of the New York Academy of Sciences 498 (Burns JJ, Rivers JM, Machlin LJ, eds) 1987.
13. Willis GC, Light AW, Gow WS. Serial arteriography in atherosclerosis. Canadian Medical Association Journal 1954;71:562-568.
14. Pauling L, Itano HA, Singer SJ, Wells IC. Sickle cell anemia, a molecular disease. Science 1949;110:543-548.
15. Mann GV, Newton P. The membrane transport of ascorbic acid. Second Conference on Vitamin C. Annals of the New York Academy of Sciences 1975;243-252.
16. Kapeghian JC, Verlangieri J. The effects of glucose on ascorbic acid uptake in heart endothelial cells: possible pathogenesis of diabetic angiopathies. Life Sciences 1984;34:577-584.
17. Dice JF, Daniel CW. The hypoglycemic effect of ascorbic acid in a juvenile-onset diabetic. International Research Communications System 1973;1:41
18. Mudd SH, Levey HL, Skovby F. Disorders of Transsulfuration. In Scriver CR, Beaudet AL, Sly WS, Valle D (eds), The Metabolic Basis of Inherited Disease 1989 McGraw-Hill:693-734.
19. Boers GHJ, Smals AGH, Trijbels FJM, Fowler B, Bakkeren JAJM, Schoonderwaldt HC, Kleijer WJ, Kloppenborg PWC. Heterozygosity for homocystinuria in premature peripheral and cerebral occlusive arterial disease. New England Journal of Medicine 1985; 313:709-715.
20. McCully KS. Homocysteine metabolism in scurvy, growth and arteriosclerosis. Nature 1971;231:391-392.
21. Fraga CG, Motchnik PA, Shigenaga MK, Helbock HJ. Jacob RA, Ames BN. Ascorbic acid protects against endogenous oxidative DNA damage in human sperm. Proceedings of the National Academy of Sciences USA 1991;88:11003-11006.
22. Pauling L. Orthomolecular psychiatry. Science 1968;160:265-271.